Chapter 9: Visualizing Cells and Their Molecules — 수업 정리본
수업 범위: 광학현미경 · 형광현미경 · 전자현미경 · 초고해상도 현미경
현미경 기법별 분해능 · 깊이 한계 비교(세포 크기 10μm)
| 기법 | 분류 | 분해능 (XY) | 관찰 가능 깊이 | 살아있는 세포 | 주요 제약·특이사항 |
|---|---|---|---|---|---|
| 명시야 (Bright-field) | 광학 | ~200 nm | 제한 없음 (깊어질수록 blur ↑) | 가능 | 염색 필요; 투명 시료 대비 낮음 |
| 암시야 (Dark-field) | 광학 | ~200 nm | 제한 없음 | 가능 | 산란광만 검출 |
| 위상차 (Phase-contrast) | 광학 | ~200 nm | 제한 없음 | 가능 (No staining) | 투명 생세포 관찰 최적; GFP 관찰 불가 |
| 형광 (Fluorescence) | 광학 | ~200 nm | 제한 없음 (out-of-focus noise) | 가능 | 특정 분자 표지 가능; blur 심함 |
| Deconvolution | 광학 + 디지털 | ~200 nm | ~40 μm (효과적) | 가능 | 회절한계 극복 불가; 컴퓨터 PSF 역계산 |
| Confocal | 광학 | ~200 nm | ~150 μm | 가능 | pinhole로 out-of-focus 제거; 아날로그 |
| Multiphoton | 광학 | ~200 nm | ~500 μm | 가능 | NIR 레이저, 광독성 낮음; 가장 깊은 광학 이미징 |
| TIRF | 광학 | ~200 nm | ~200 nm (evanescent field) | 가능 | 세포막 표면 극히 제한; 단일 분자 관찰 가능 |
| SIM | 초고해상도 | ~100 nm | 수십 μm (3D 가능) | 어려움 | 모든 형광 dye 사용 가능; 약 2배 향상 |
| STED | 초고해상도 | ~20 nm | confocal 수준 | 어려움 | Photoswitchable probe 필수; depletion beam (도넛형) |
| STORM) | 초고해상도 | ~20 nm | ~200 nm (TIRF 조합 시) | 어려움 | 수만~수십만 cycles; 긴 촬영 시간 |
| TEM | 전자현미경 | 실용 ~0.05 nm / 생물 ~1 nm | 수십 nm (초박절편) | 불가 | 이론적 0.002 nm; 진공, 중금속 염색 필요 |
| SEM | 전자현미경 | ~10 nm | 표면만 | 불가 | 3D 표면 이미지; 내부 구조 관찰 불가 |
| Cryo EM | 전자현미경 | 원자 수준(~nm) | 수백 nm (FIB lamella) | 불가 | 비결정질 동결; 2017 노벨화학상 |
| AFM | 기타 | 수직 0.1 nm / 수평 1 nm | 표면만 | 가능 | 힘 측정; 살아있는 세포 표면 관찰 가능 |
깊이 한계 요약 (깊은 것 → 얕은 것)
Multiphoton 500 μm > Confocal 150 μm > Deconvolution 40 μm > 형광/위상차 (blur 있으나 제한 없음) > TIRF / SMLM 200 nm ≈ TEM 초박절편 수십 nm > SEM · AFM (표면만)
현미경과 노벨상
| 인물 | 업적 | 수상 |
|---|---|---|
| Frits Zernike (1888–1966) | 위상차 현미경 발명 | 1953 물리학상 |
| Ernst Ruska (1906–1988) | 전자현미경 개발 | 1986 물리학상 |
| Gerd Binnig / Heinrich Rohrer | 주사형 터널링 현미경(STM) 개발 | 1986 물리학상 공동 수상 |
| GFP 발견·개발 관련 연구자 | GFP의 발견과 개발 | 2008 화학상 |
| Eric Betzig / Stefan W. Hell / William E. Moerner | 형광 분자를 이용한 초고해상도 광학현미경 | 2014 화학상 |
| Jacques Dubochet / Joachim Frank / Richard Henderson | Cryo-electron microscopy 개발 | 2017 화학상 |
관찰 규모 개요
현미경 별 관찰 규모
Link to original교과서 도식
- 광학현미경: naked eye(1 cm) ~ 200 nm(회절한계) 범위
- 전자현미경: ~1 nm까지 관찰 가능
- 초고해상도(Super-resolution): 200 nm 장벽 극복 가능
이 챕터의 세 가지 큰 주제
광학 현미경 / 염색 & GFP / 전자 현미경
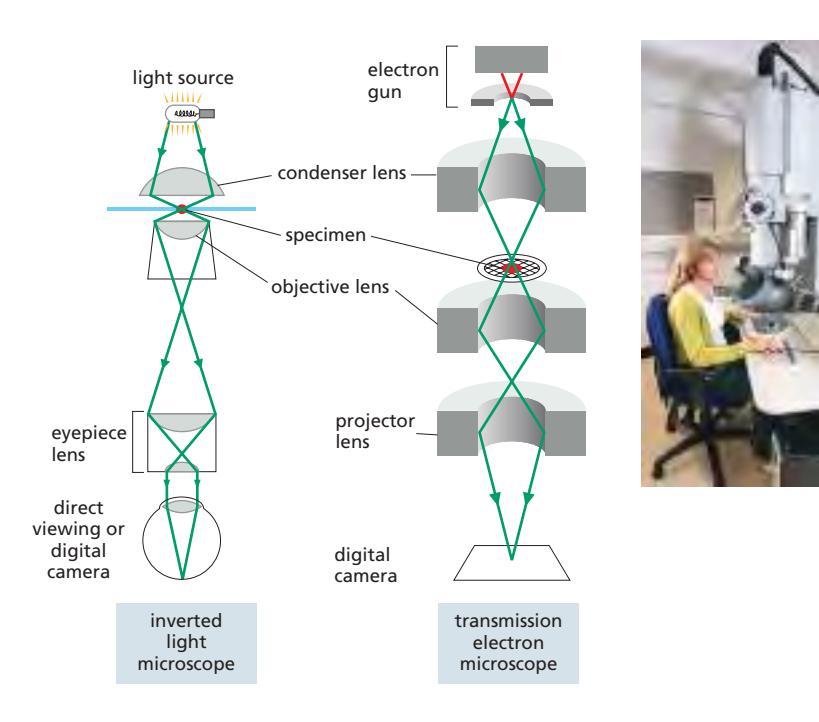
1. 광학현미경 (Optical Microscope)
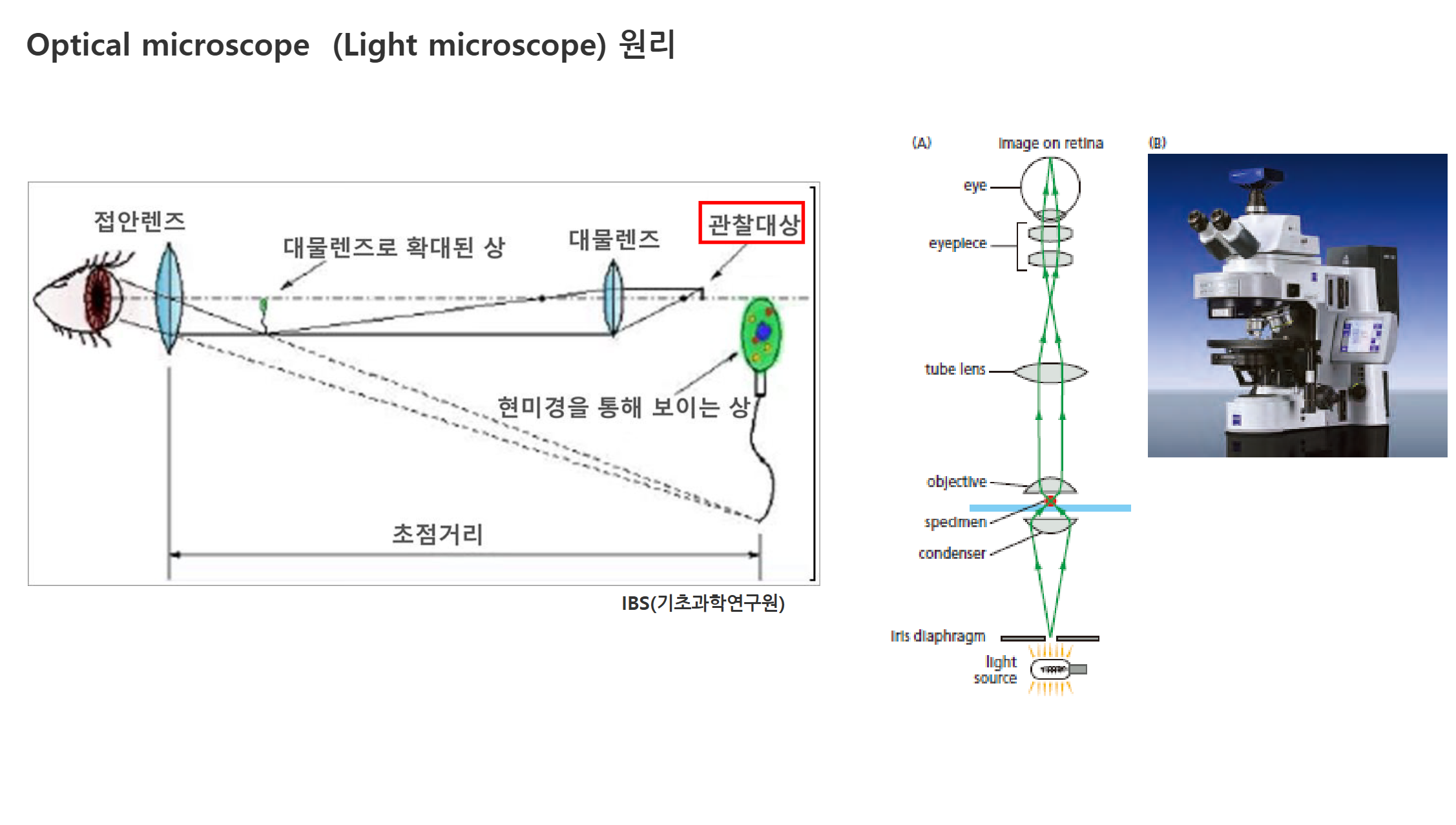
원리 요약
- 접안렌즈(eyepiece): 대물렌즈가 만든 상을 한 번 더 확대
- 대물렌즈(objective): 관찰 대상을 1차 확대
- 광원 → iris diaphragm → condenser → specimen → objective → tube lens → eyepiece
- Dark field(암시야): 굴절된 빛만 감지 / Bright field(명시야): 직진광과 약간 산란된 빛까지 감지
분해능 (Resolution)
→ limit of resolution(분해능) / diffraction limit
- : 구분 가능한 최소 거리
- : 빛의 파장
- : 렌즈의 빛 모음 정도
가시광선(400–700 nm) → 분해능 한계 약 200 nm
- 회절한계(Diffraction limit): 물리 법칙에 의한 이론적·물리적 분해능의 한계
- 완벽한 렌즈를 써도 극복 불가능
- 빛의 세기, 시료 두께와는 무관 (파장·개구수에만 의존)
- 초고해상도 현미경은 다른 물리적 원리를 이용해 이를 극복
2. 광학현미경 대비 (Contrast) 형성 원리
일반 광학현미경으로 세포 관찰 시 → 투명 유리판에 투명 젤 같은 느낌 → 대비 형성 필수
3가지 대비 형성 방법
| 방식 | 원리 | 현미경 종류 |
|---|---|---|
| (A) 흡수 차이 | 염색약이 특정 파장을 흡수 (예: 적색 염색약 → 초록/파랑 흡수, 빨강 통과) | 명시야(Bright-field) |
| (B) 산란 | 비스듬한 입사광 → 세포 성분에 의해 산란된 빛만 대물렌즈로 수집 → 어두운 배경에 밝은 이미지 | 암시야(Dark-field) |
| (C) 위상 차이 | 빛의 위상이 세포의 더 두껍거나 밀도가 높은 부분을 통과하면서 변함 → 보강/상쇄간섭 → 밝기 차이로 변환 | 위상차(Phase-contrast) |
3. 위상차 현미경 (Phase Contrast Microscopy)
→ phase contrast microscopy(위상차 현미경)
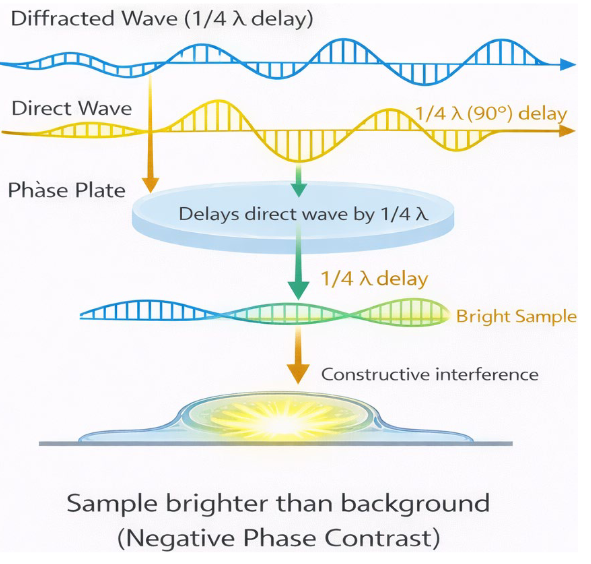
- 무색 투명한 시료도 관찰 가능 (Live cell imaging / No staining)
- 빛의 위상차를 명암 차이로 변환: 세포의 밀도 차이로 생긴 보이지 않는 빛의 시간차를 특수 렌즈로 눈에 보이는 밝기 차이로 증폭
- 형광(fluorescence)을 사용하지 않으므로 GFP 관찰 불가
- 지엽) 1935년 F.제르니커 개발, 1953년 노벨 물리학상
원리 — Negative Phase Contrast (강의에서 이것만 언급)
- 세포를 통과한 회절광: 굴절률 차이로 인해 λ/4 지연 발생
- 직진광: 위상판(Phase Plate)에 의해 λ/4 추가 지연
- → 두 빛의 위상이 같아져 보강간섭 → 시료 부분이 밝게 보임
Positive phase contrast: 직진광 3λ/4 지연 → 상쇄간섭 → 시료 어둡게 보임
구성요소
- 위상차용 대물렌즈 (렌즈에 PH: Phase 표기)
- 위상차 콘덴서(Phase Contrast Condenser)
4. 조직 고정 및 절편 (Fixation & Sectioning)
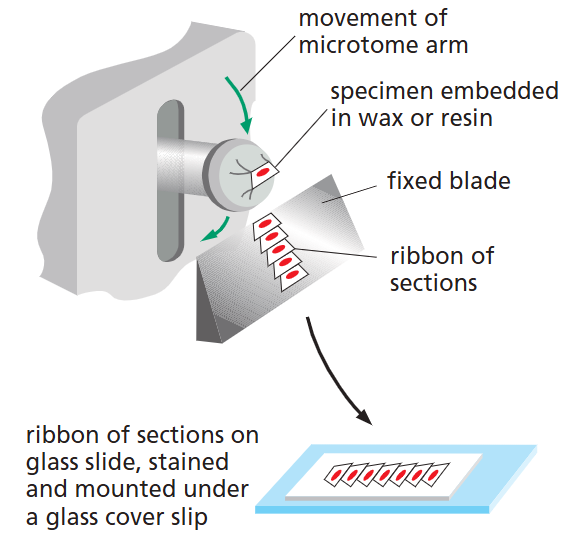
조직은 너무 두꺼워 고해상도 직접 검사 불가 → 절편(section) 필요 (임상에서 중요한 부분)
준비 과정
고정(Fixation) → 포매(Embedding) → 절단(Sectioning) → 염색(Staining)
- 고정(Fixation): Glutaraldehyde (단백질 교차결합) → 분해 방지, 구조 보존
- 고정하지 않으면 모두 분해됨
- 포매(Embedding): 파라핀 또는 레진을 조직에 침투 → 고체 블록 형성
- 고정 후에도 조직은 부드럽고 연약해서 포매 필요
- 절단: 마이크로톰(Microtome)으로 절단 → 일반적으로 0.5–10 μm 두께
- 염색: 절단된 절편에 염색 필수
- DNA/RNA/산성단백질: H&E 염색 (hematoxylin & eosin)
- 다양한 항체로 단백질 발현 비교
- 특정 단백질 위치: 형광 probe + marker (GFP/RFP) 이용
5. 형광현미경 (Fluorescence Microscopy)
→ 000_Fluorescence Microscopy(형광현미경)
형광(Fluorescence) 원리
형광 분자(형광색소, fluorochrome):
- 특정 파장의 빛(광자)을 흡수 → 전자가 excited state로 상승
- 전자가 ground state로 돌아오면서 더 긴 파장(낮은 에너지)의 빛 방출 = 형광
형광현미경 필터 시스템 (3단계)
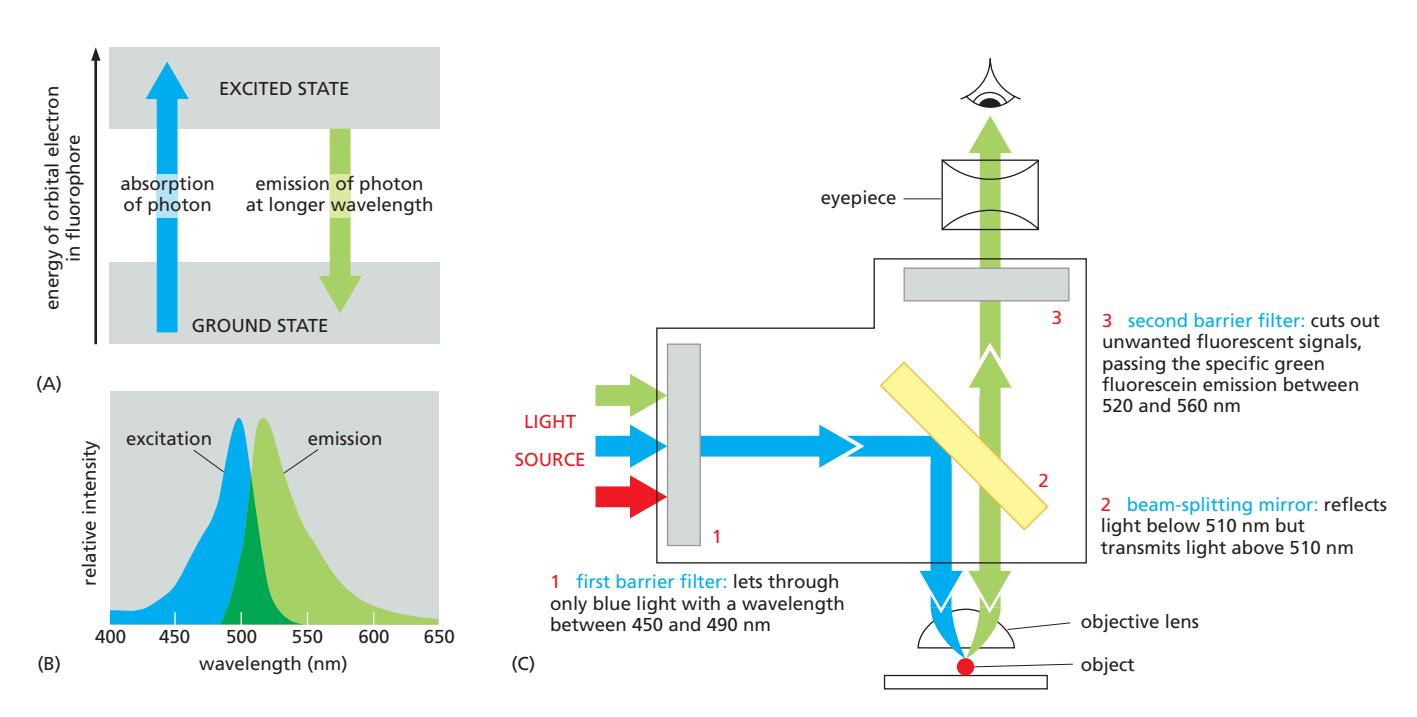
1. Excitation filter (1st barrier filter)
- 특정 파장만 통과 (예: 450~490 nm 파란빛만)
2. Beam-splitting mirror (Dichroic mirror)
- 510 nm보다 짧은 파장(파란빛) → 반사
- 510 nm보다 긴 파장(형광 emission) → 통과
3. Second barrier filter (Emission filter)
- 520~560 nm (형광 emission)만 통과
주요 형광 염료 조합
| 색 | 염료 예시 |
|---|---|
| 파랑 | DAPI (DNA 염색) |
| 초록 | GFP, FITC |
| 빨강 | Rhodamine B, Cy3, RFP, Cy5 |
GFP, FITC, YFP는 방출광이 겹쳐 동시에 사용 불가. 보통 DAPI(파랑) + GFP(초록) + Rhodamine/Cy3(빨강) 조합 사용. 노란색은 안 씀 — 녹색 + 붉은색인지 고유한 단백질인지 판별 불가.
Antibody를 이용한 특정 분자 검출
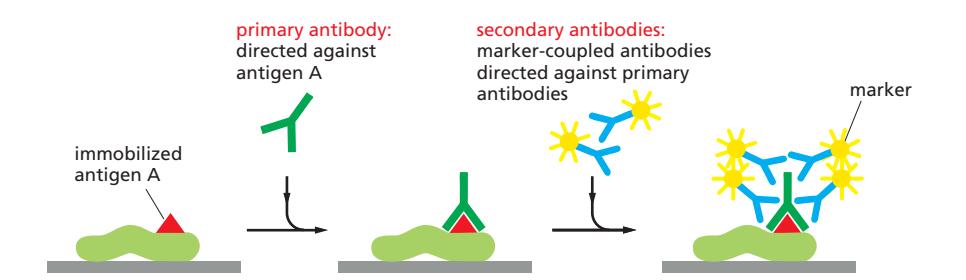
Direct vs Indirect Immunocytochemistry
| 방법 | 원리 | 특이도 | 민감도 |
|---|---|---|---|
| Direct (직접) | 1차 항체에 형광 직접 결합 | 높음 (specific) | 낮음 |
| Indirect (간접) | 1차 항체 + labeled 2차 항체 | 낮음 | 높음 (sensitive) |
- 1차 항체(Primary antibody): 타겟 항원에 결합
- 2차 항체(Secondary antibody): 1차 항체에 결합; secondary antibodies로 검출하면 더 강한 signal 획득
- Indirect method가 더 sensitive(민감)하지만 덜 specific함
- 여러 2차 항체 분자가 각 1차 항체를 인식 → signal 증폭 but nonspecific 형광 noise 발생 가능
immunocytochemistry의 단점
살아있는 세포에 antibody를 집어넣는 침습적 과정에서 세포가 죽음 → 살아있는 세포 관찰 불가능 → 대안: GFP fusion protein
6. GFP (Fluorescent Protein Tagging)
→ GFP(Fluorescent Protein Tagging)
GFP 특징
- 자체적으로 형광을 띠는 단백질 (외부 형광 염료 불필요)
- Barrel 구조 (11개의 β-strand) 내부에 chromophore 보호
- 번역 직후에는 형광 없음 → 약 1시간 이내 자가촉매적 번역 후 변형으로 성숙
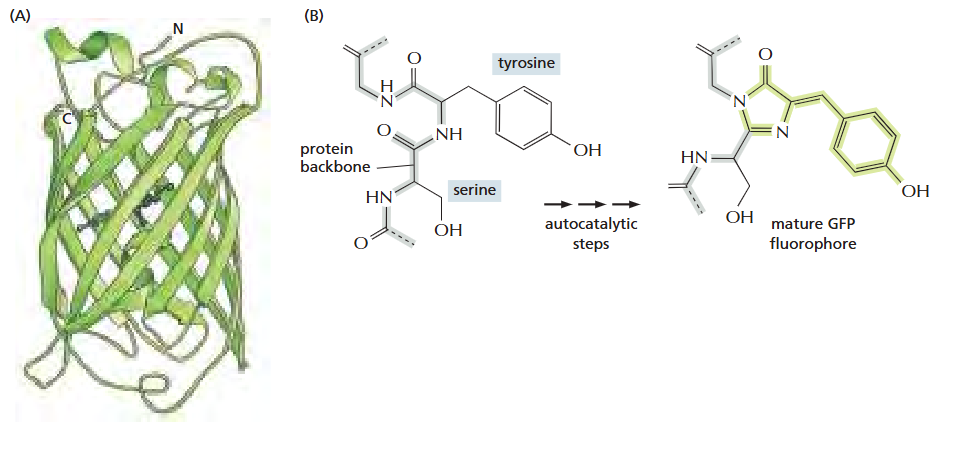
GFP 활용 방법
1. Reporter molecule: GFP 유전자를 관심 유전자의 promoter 하에 배치 → 유전자 발현 패턴 시각화
(A) 초파리 실험 예시: GFP 유전자를 초파리 신경세포 특이적 promoter에 연결 → 신경세포만 형광 발현
2. GFP Fusion Protein (가장 중요):
ATG-XXXXXX(target 단백질)----GFP(형광)
↓
Fusion protein
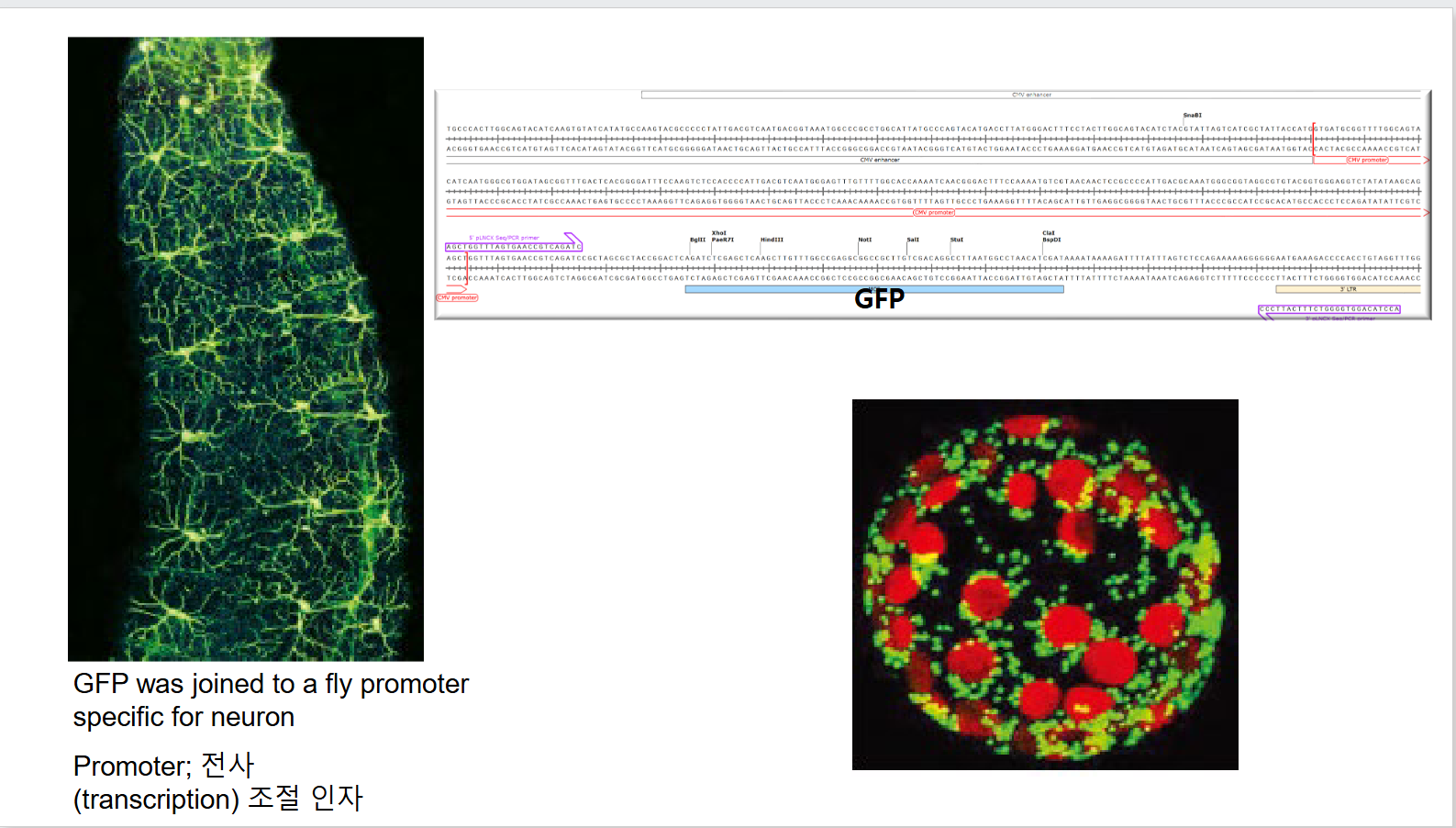
- 살아있는 생물에서 단백질의 **distribution(분포)**과 **dynamics(역학)**를 표시하는 가장 명확한 방법
- GFP fusion protein이 untagged protein과 기능적으로 동등함을 증명해야 함 (예: mutant rescue로 확인)
- 대부분의 경우 GFP fusion protein이 원래 단백질과 동일한 방식으로 작동
3. Organelle targeting: GFP에 peptide location signal 추가 → 특정 organelle 형광 표지
7. 3D 이미지 획득
PSF와 Image Deconvolution
→ PSF(point spread function) / deconvolution
- PSF (Point Spread Function): 빛의 퍼짐 정도 — 점 광원이 렌즈를 통과할 때 형성되는 3D blurred image (Gaussian distribution에 근사)
- 광학현미경 분해능 한계, 샘플 두께, 초점 부정확성 등으로 이미지 왜곡 발생
- Image deconvolution (이미지 복원): 컴퓨터 알고리즘으로 PSF 신호 최소화, noise 제거 → 흐려진 이미지 복원
- 추가 수업내용: 컴퓨터가 계산해서 만든 가상의 뿌연 이미지와 실제로 현미경으로 관찰된 진짜 뿌연 이미지를 비교 → 차이가 작으면 진짜 모습
- Diffraction limit에 의해 여전히 제약 (없는 정보를 만들어내지는 못함)
- 표본 깊이 약 40 μm까지 효과적
Computed Tomography 개념
- 다른 각도에서 찍은 이미지를 결합 → 이미지 재구성 (CT scanner와 동일한 원리)
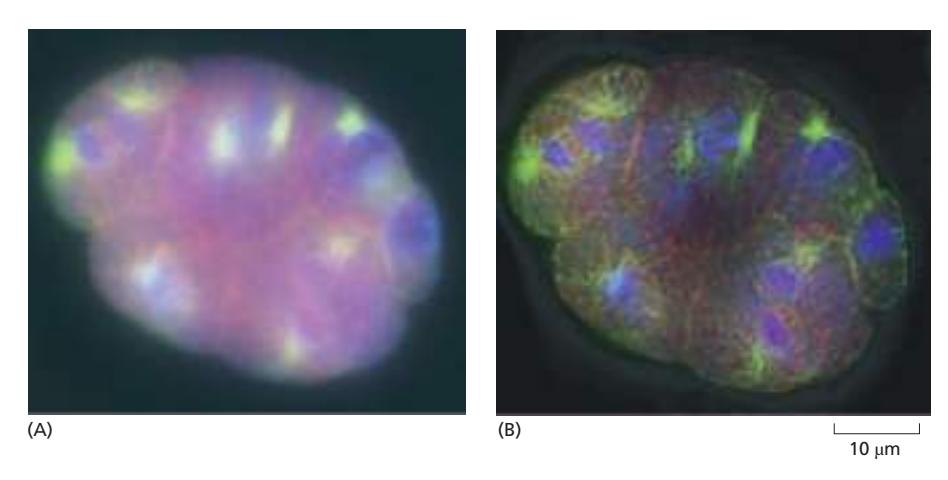
8. 공초점 현미경 (Confocal Microscope)
일반 형광현미경: 시료 전체에 빛 조사 → out-of-focus 빛으로 blur Confocal: 매우 작은 선택 영역만 선택적으로 빛 조사
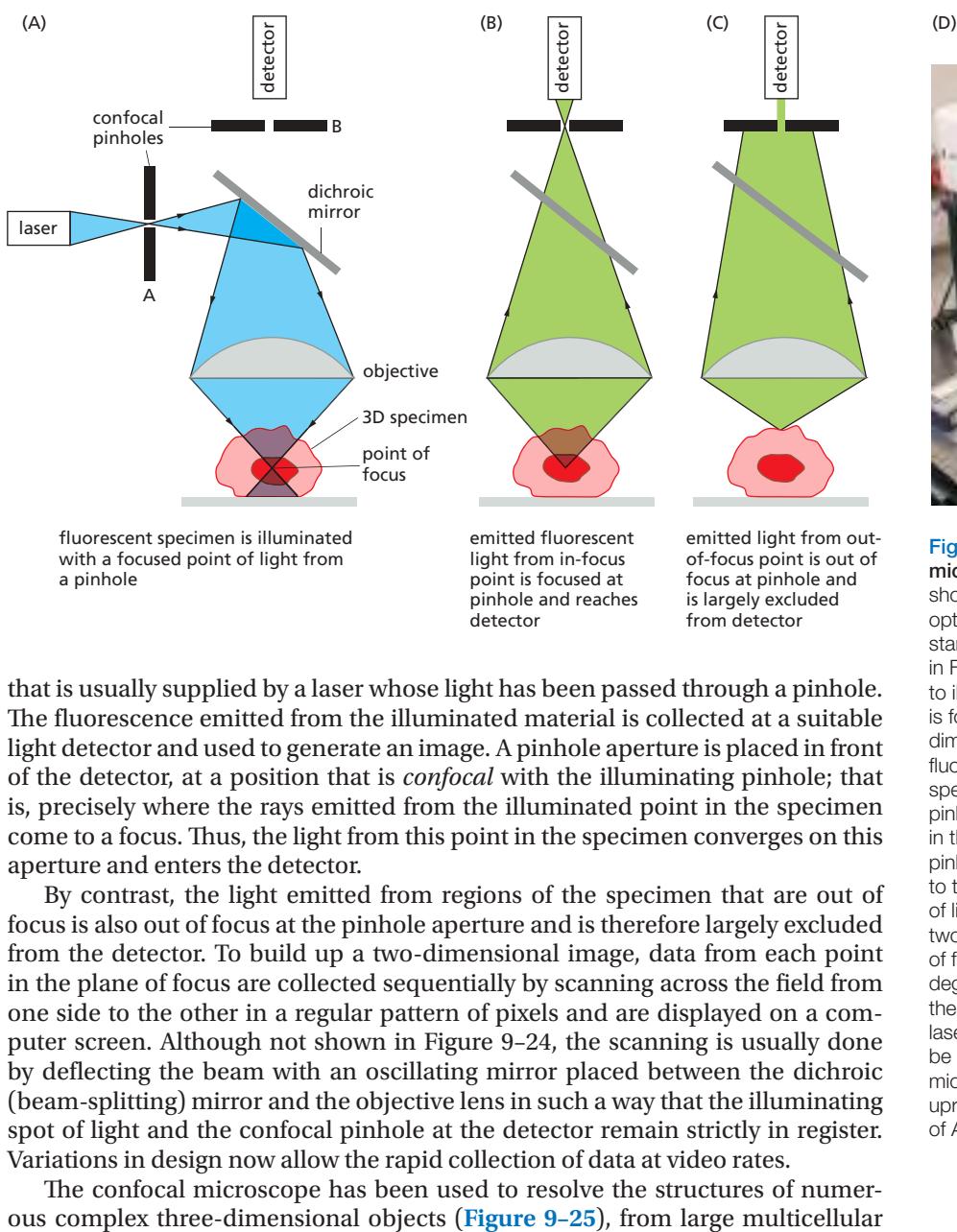
원리
- Laser → pinhole A 통과 → 표본의 단일 focal point에 집속
- 그 점에서 방출된 형광 → confocal pinhole B에 집속 → detector 도달
- 초점 밖(out-of-focus) 빛은 pinhole에서 초점 안 맞음 → detector 대부분 제외
- 다른 깊이에서 촬영한 optical section을 쌓아 3D 이미지 재구성
특징
- 아날로그 방식 (deconvolution은 디지털 방식)
- 최대 약 150 μm 깊이까지만 이미지 획득 가능
그럼에도 noise가 발생하는 이유 (→ Multiphoton의 필요성)
- 빛의 경로에 있는 시료에도 형광이 발생 → 그 빛이 pinhole 가장자리로 소량 들어옴
- out-of-focus 빛 중에 PSF의 꼬리부분에 해당하는 빛이 pinhole로 들어옴
- out-of-focus 빛이 시료 내에서 산란·굴절되며 pinhole로 들어옴
→ 이를 해결하기 위한 대안 = Multiphoton Microscopy
9. Multiphoton Microscopy
배경
- 형광 분자를 들뜨게 하려면 보통 짧은 파장(고에너지) 필요
- 짧은 파장(청색/자외선)은 산란과 흡수가 커서 깊이 침투 불리
- **근적외선(NIR, 긴 파장)**은 산란이 덜해 더 깊이 침투 가능
원리
- 근적외선(NIR) 레이저로 **두 광자(two-photon)**를 femtosecond 이내에 동시 흡수 → 형광 들뜸
- 특수 장비로 초점 지역에만 two-photon이 도달하게 함
- 광자 밀도가 높은 초점 지역에서만 의미있는 흡수가 일어남 → 그 외 지역은 형광 발생 없음 → noise 없음
장점
- 더 깊이 침투: NIR은 산란이 덜해 표본 내 ~500 μm 깊이까지 이미지 획득 가능
- 낮은 광독성: 적외선 레이저가 가시광보다 세포 손상 적음
- Background noise 감소 (confocal의 noise 문제 해결)
10. 살아있는 세포의 단백질 역학 (Protein Dynamics in Living Cells)
→ 000_Protein Dynamics in vivo MOC
(1) FRET — Fluorescence Resonance Energy Transfer
→ FRET
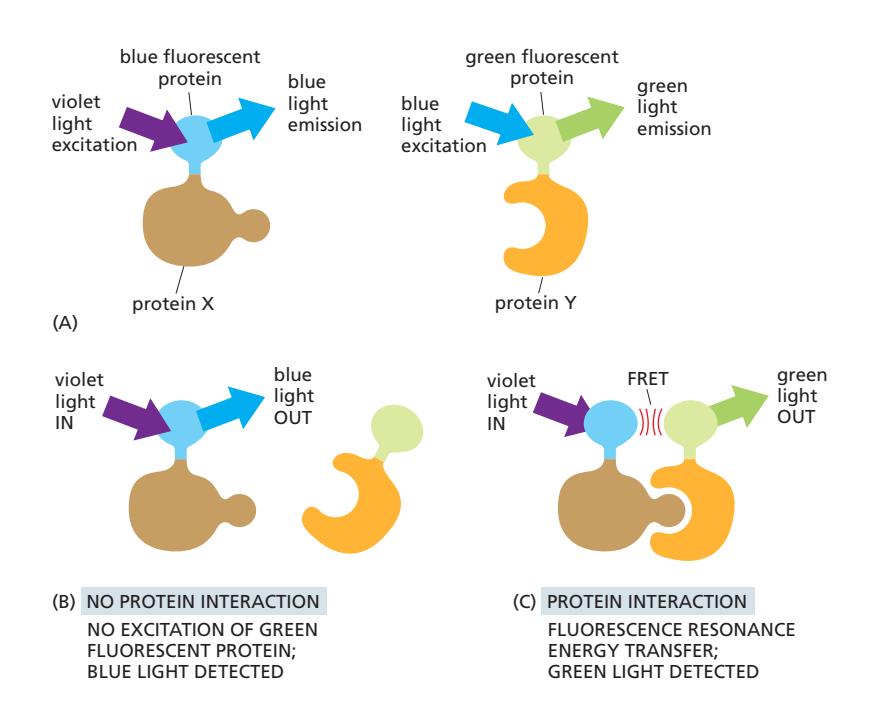
- 관심 있는 두 분자를 각각 다른 fluorochrome으로 표지
- 선택 기준: donor의 emission spectrum과 acceptor의 absorption spectrum이 **overlap(겹침)**해야 함
- 두 단백질이 약 1–5 nm 이내로 근접 시, donor의 형광 에너지가 acceptor로 **비방사적(nonradiatively)**으로 전달 (resonance)
- 상호작용 없으면: violet 여기 → blue emission만 / 상호작용 있으면: violet 여기 → green emission 검출
- FRET 효율은 거리의 6제곱에 반비례(1/r⁶) → 극도로 민감
- 단백질-단백질 상호작용 실시간 모니터링에 활용 (살아있는 세포에서)
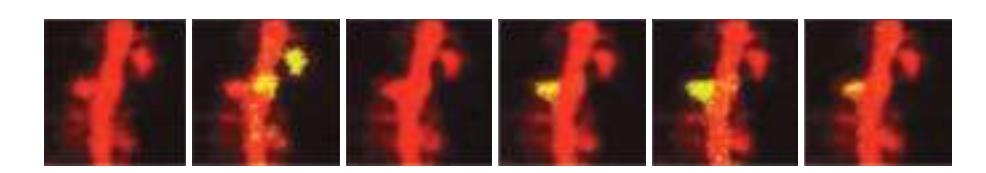
(2) Photoactivation (광활성화)
형광 vs 광활성화
형광: 활성화 과정 없이 특정 파장의 빛을 받으면 형광 발생 광활성화: 빛에 의해 화학 구조가 변해 새로운 형광 특성을 가짐 → 단일 분자 추적 가능
- 빛에 의해 화학 구조가 변해 새로운 형광 특성을 가짐 — 원하는 순간, 원하는 위치에 형광
- 과정:
- 활성화: 강한 빛 (자외선 or 청색광)으로 photoactivatable probe 활성화
- 확인: 형광을 확인할 수 있는 빛으로 이동 추적
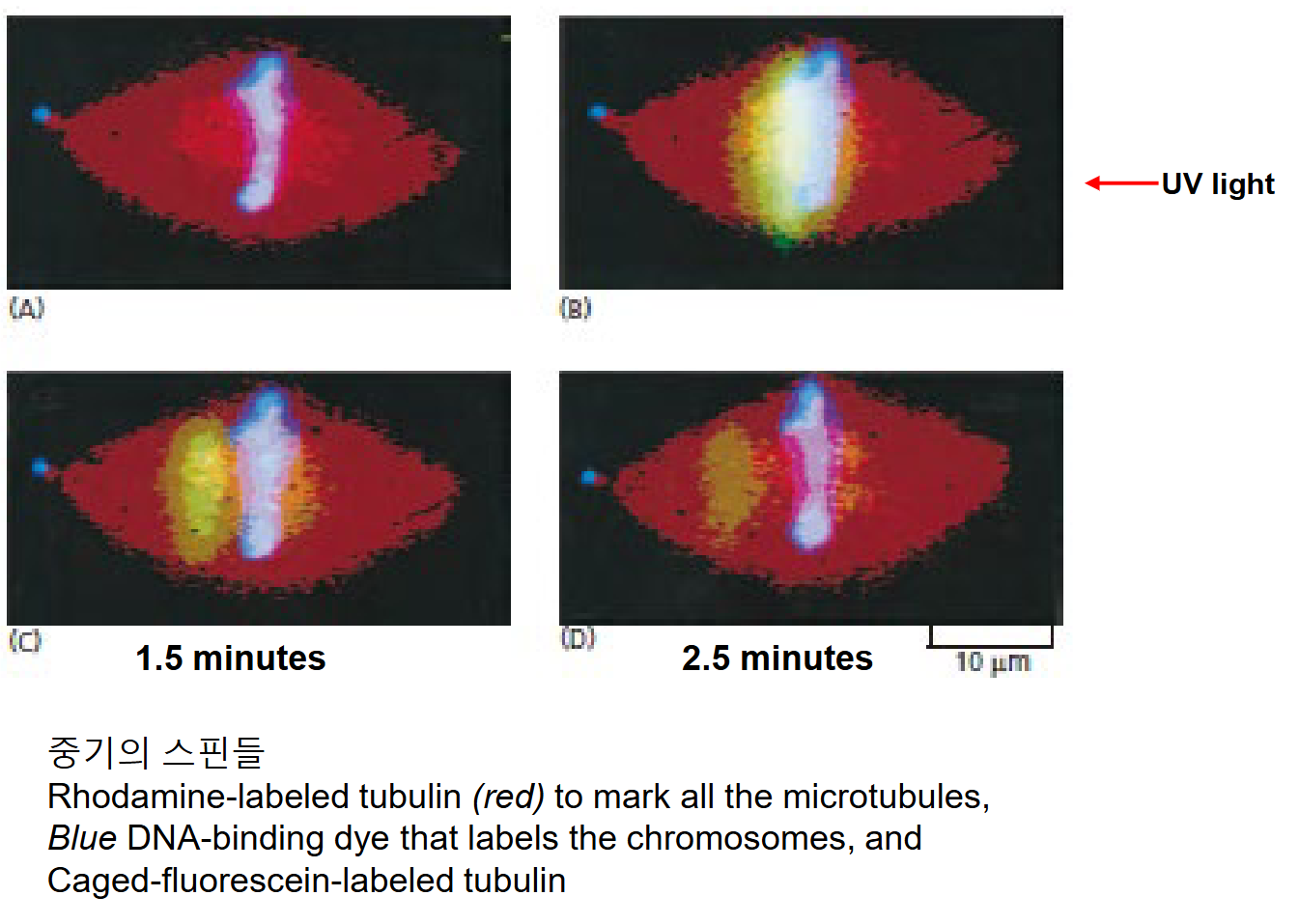
가운데 tubulin 단백질들에만 UV로 광활성화 → 2.5분간 광활성이 줄어드는 동시에 단백질의 이동을 관측 → Spindle dynamics 확인
(3) FRAP — Fluorescence Recovery After Photobleaching
→ FRAP
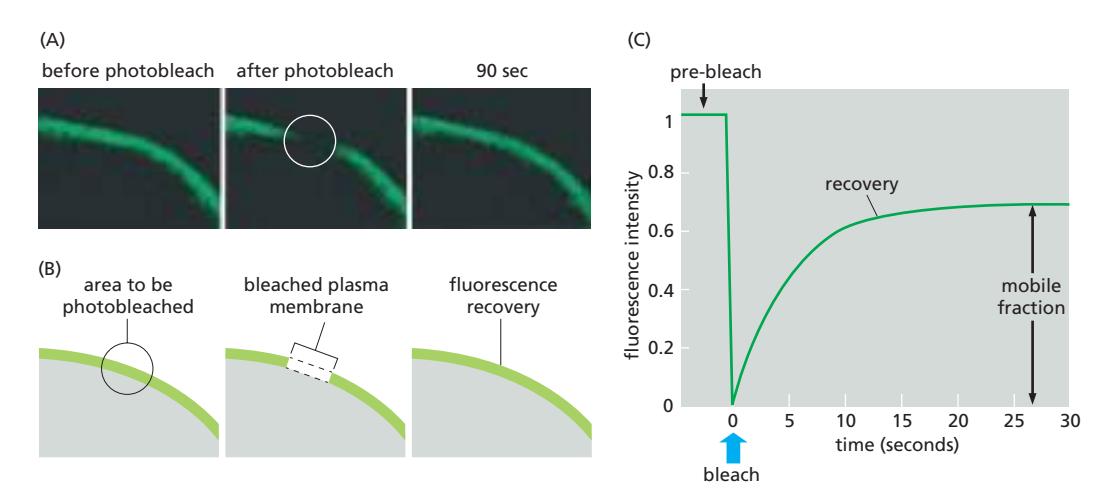
- 특정 영역에 강한 레이저를 조사해 형광을 photobleaching(표백)
- 표백된 영역으로 주변의 형광 분자들이 이동해 들어오면서 형광 회복 관찰
- population level 측정 (단일 분자 추적이 아님)
- 측정 가능한 정보:
- Diffusion coefficient (확산 계수): 회복 곡선의 기울기
- Mobile fraction (이동 가능 분획): 최종 회복 정도
- Immobile fraction: 회복되지 않은 비율 (고정되거나 매우 느리게 이동하는 분자)
11. 세포 내 이온 농도 측정
→ Aequorin=Light Emitting Indicator
Aequorin: Ca²⁺가 있을 때만 청색광을 방출하는 발광 단백질 (형광이 아님)
- 0.5~10 μM 범위의 Ca²⁺ 농도 변화에 반응
- 발광은 형광보다 신호가 약해 실험적으로 형광 물질이 더 많이 사용됨
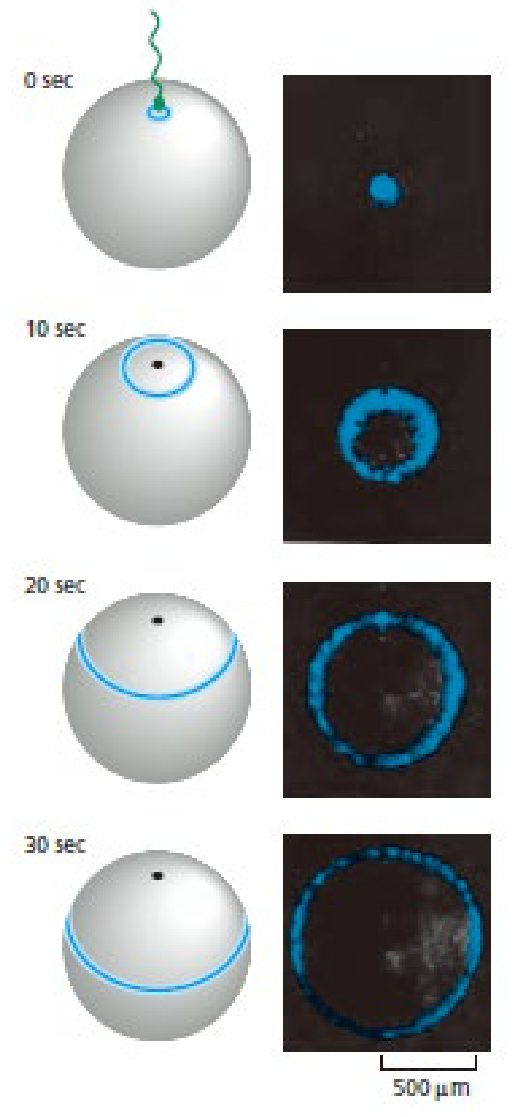
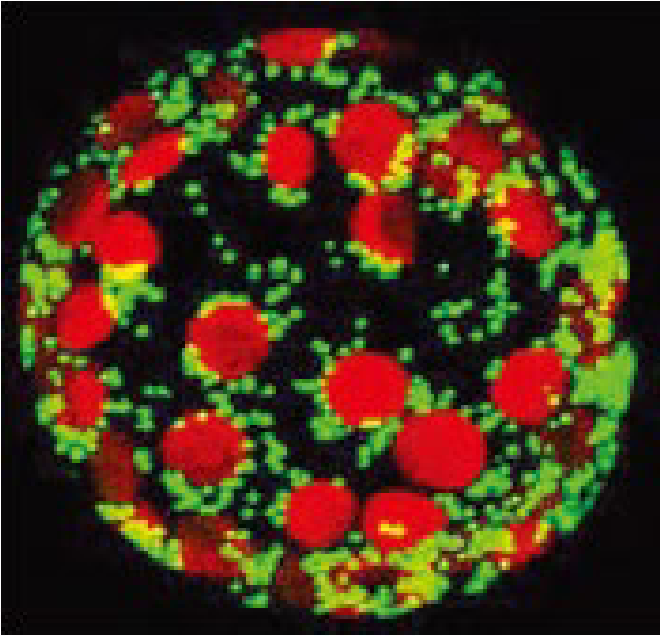
형광 Ca²⁺ indicator: Fluorescent Biosensors for Cell Signaling
- 모든 indicator가 Ca²⁺에 결합하는 게 아니라, 결합한 것과 안 한 것이 나뉘어 농도에 의해 색 차이 형성

- 빨간색: 신호 약함 / 노란색: 신호 강함 (Purkinje 세포 예시)
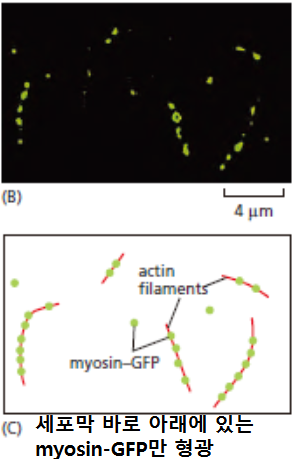
12. TIRF — Total Internal Reflection Fluorescence Microscopy
→ TIRF(Total Internal Reflection Fluorescence Microscopy)
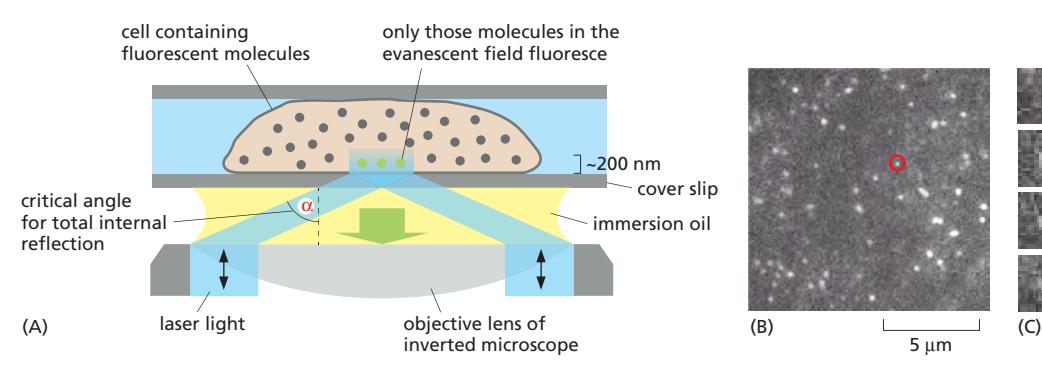
원리
- 레이저를 임계각 이상으로 cover-slip에 입사 → 전반사(Total Internal Reflection) 발생
- 전반사에도 불구하고 **evanescent field(감쇠장, 소멸파)**가 표면 너머로 약 200 nm까지만 확장
- → cover-slip에 부착되었거나 표면에 매우 가까운 분자들만 여기
특징
- **세포 두께(10 μm)의 약 1–2%**인 ~200 nm 깊이만 관찰
- 세포막 바로 아래 단백질 같은 막 근처 구조 관찰에 최적
- Background 대폭 감소 → noise 없음, 단일 분자 관찰 가능
- 광독성 감소 / 광표백 최소화: 제한된 영역만 조명
강의노트: myosin, actin filament 관찰
13. AFM — Atomic Force Microscopy (원자력 현미경)
→ AFM(Atomic Force Microscopy)
힘(force)을 이용해 **표면 구조와 물성(topology·마찰·정전 상호작용·도전률·자력)**을 image화
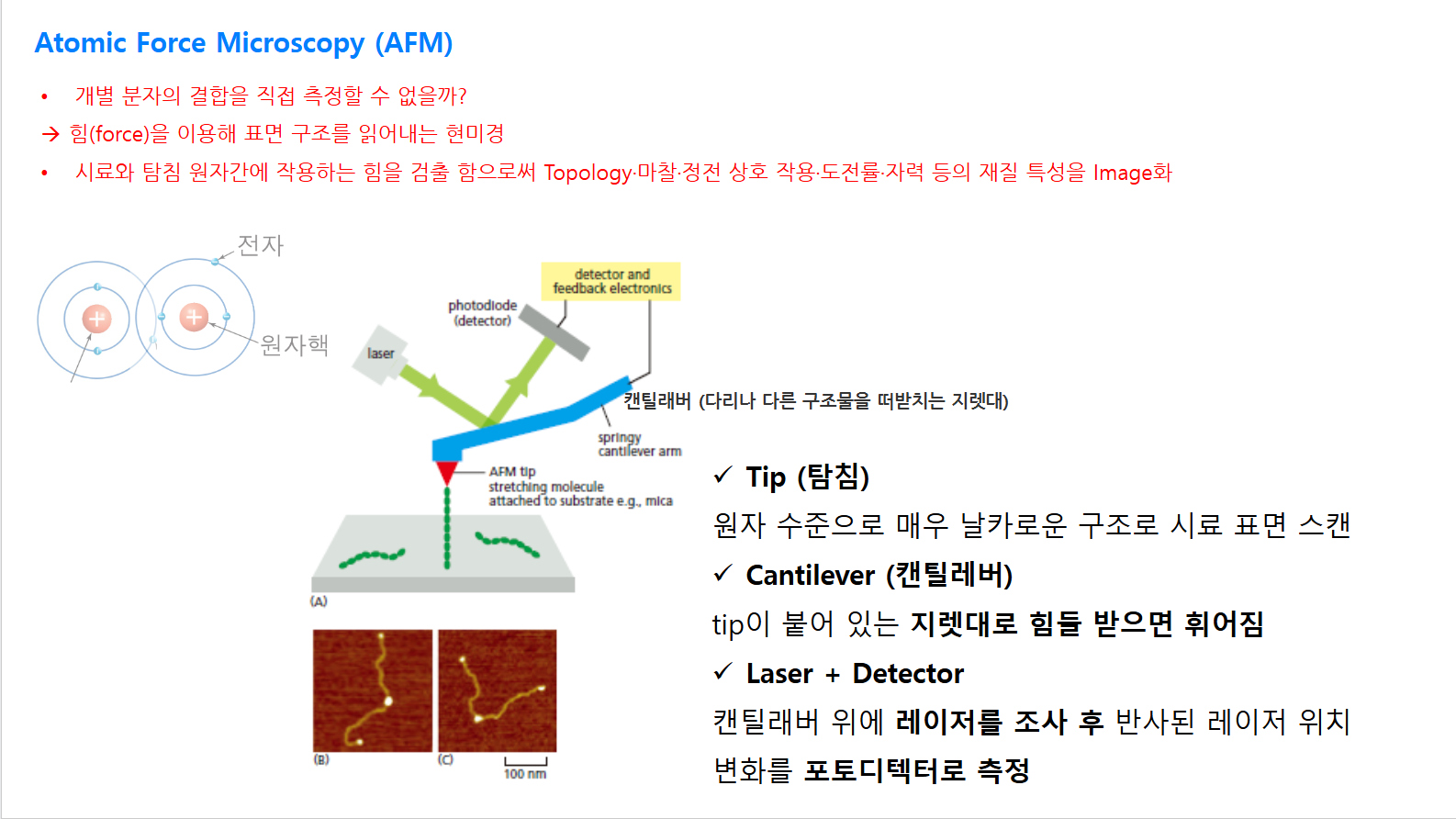
구성요소
- Tip (탐침): 원자 수준으로 매우 날카로운 구조로 시료 표면 스캔
- Cantilever (캔틸레버): tip이 붙어 있는 지렛대 → 힘을 받으면 휘어짐
- Laser + Detector: 캔틸레버 위에 레이저 조사 후 반사된 레이저 위치 변화를 **photodiode(detector)**로 측정
응용
- 생물학적 시료(DNA, 단백질, 세포) 관찰
- 예: DNA 가는 선 / MUTs 밝은 점 → DNA damage가 있는 부분에만 MUTs 단백질 검출
- 해상도: 수직 0.1 nm, 수평 1 nm
14. 초고해상도 현미경 (Super-resolution Microscopy)
일반 광학현미경의 회절한계(~200 nm)를 극복하는 기술들 — 모두 광학현미경에 속함!
(1) SIM — Structured Illumination Microscopy
→ SIM(Structured Illumination Microscopy)
전자현미경보다 나은 광학현미경이 나왔다!!!

원리:
- 모아레 패턴(Moiré pattern): 두 개의 주기적인 패턴이 겹쳐질 때 생성되는 간섭 무늬
- 회절한계에 의해 얻을 수 없는 샘플의 고분해능 정보가 관찰 가능한 저분해능 모아레 무늬에 반영
- Known 레이저 패턴과 Unknown 샘플 구조가 결합 → moiré 패턴 생성
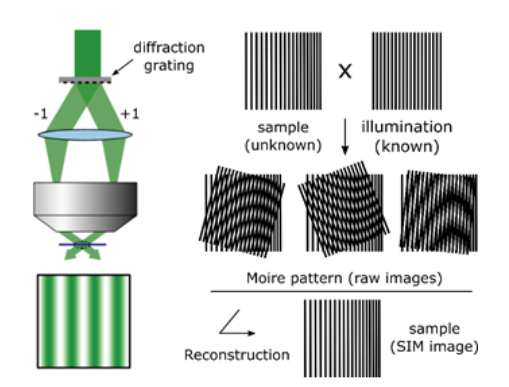
- 조명 패턴 각도를 바꾸면서 여러 장 촬영 → 컴퓨터로 역계산 → 숨겨진 고해상도 구조 복원
특징:
- 분해능: ~100 nm (conventional의 약 2배 향상)
- 모든 fluorescent dye/protein 사용 가능 (versatile)
- 완전히 회절한계를 벗어나진 못함 (약 2배 향상에 그침)
(2) STED — Stimulated Emission Depletion Microscopy
→ STED(Stimulated Emission Depletion)
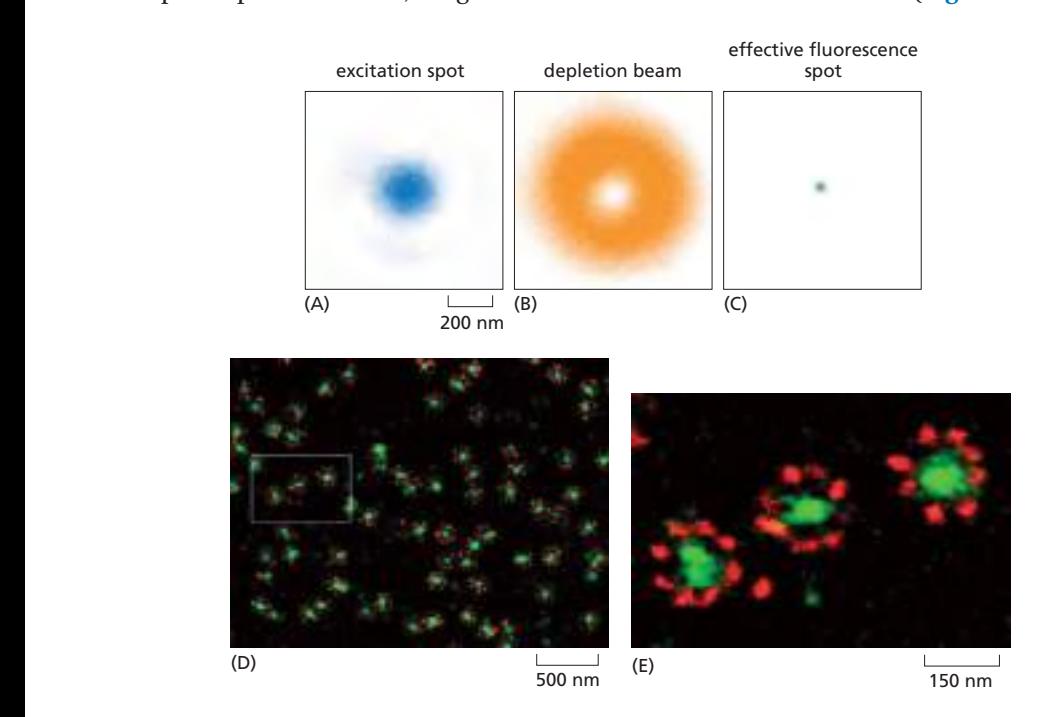
원리:
- Excitation laser: 형광 여기 spot 생성
- Depletion laser (도넛 모양, doughnut shape): spot 주변의 형광 분자를 stimulated emission으로 ground state로 되돌림 (photobleaching 아님! photoactivation 아님!)
- → 오직 beam 정중앙의 극소 영역에서만 형광 발생 → PSF 크기 감소
- Confocal과 유사하게 point-by-point scanning
특징:
- 분해능: ~20 nm (conventional의 약 10배 향상)
- Photoswitchable fluorescent probes 필요
- 매우 가까이 있는 분자들이 두 가지 다른 states(형광/어두운) 중 하나에 있도록 보장 → 회절 한계 극복
(3) SMLM — Single-Molecule Localization Microscopy
→ SMLM (Single-Molecule Localization Microscopy)
대표: PALM (Photo-Activated Localization Microscopy)
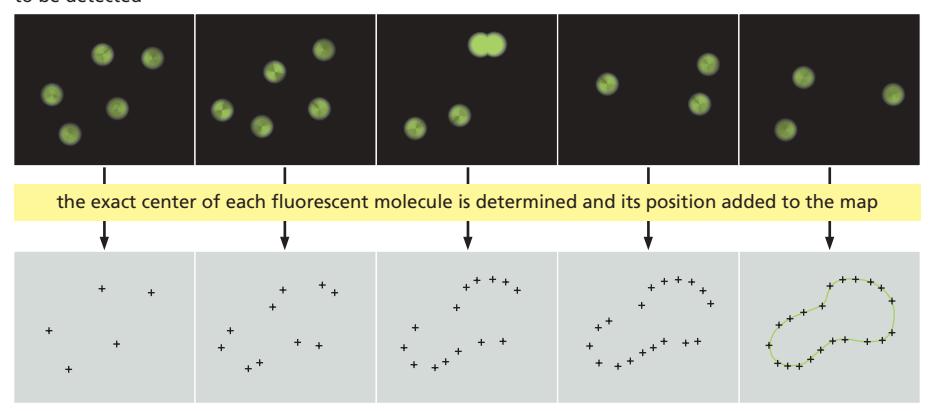
3단계 원리:
1. Isolation (분리): 모든 형광 분자를 한 번에 켜지 않고, 아주 적은 수의 분자만 무작위로 형광 2. Localization (위치 결정): PSF 때문에 희미하게 보여도 정 가운데 부분은 분자의 중심 → 중심점(Center) 확인 3. Reconstruction (재구성): 반복하여 얻은 수만 개의 점 데이터를 하나의 이미지화
특징:
- 분해능: ~20 nm (conventional의 약 10배 향상)
- Photoswitchable labels (photoactivated / photoconvertible / photoswitchable) 필요
- 긴 acquisition time (수만~수십만 cycles)
- TIRF microscope와 조합해서 사용
초고해상도 기술 비교표
| 기술 | 분해능 | 원리 | 특이사항 |
|---|---|---|---|
| SIM | ~100 nm | Moiré pattern + 역계산 | 모든 dye 가능, 빠름 |
| STED | ~20 nm | PSF 축소 (depletion beam) | Photoswitchable probe 필요 |
| SMLM (PALM) | ~20 nm | 단일 분자 위치 결정 후 재구성 | 매우 긴 촬영 시간 |
15. 전자현미경 (Electron Microscope)
개요
| 종류 | 원리 | 특징 |
|---|---|---|
| TEM (투과 전자현미경) | 전자빔을 시료를 통과시켜 이미지 생성 | 내부 구조, 초고분해능 |
| SEM (주사 전자현미경) | 전자빔이 시료 표면을 탐침하여 이미지 생성 | 표면 3D 이미지 |
| Cryo EM | TEM의 한 종류; 시료를 초저온에서 이미지 획득 | 생체 분자구조 분석, 2017 노벨화학상 |
TEM (Transmission Electron Microscope)
→ TEM(Transmission Electron Microscope)
빛 대신 전자를 사용 — 전자도 파동의 성질을 가짐
- ==전자의 파장은 속도가 빠를수록 짧아짐 (드브로이 파장: λ = h/mv)
- 100–300 keV 가속 시 파장 0.0037 nm (가시광선 대비 약 10만배 짧음)
- 이론적 분해능: ~0.002 nm (파장의 약 절반)
- 실용적 분해능: ~0.05 nm (렌즈 수차 때문)
- 생물학적 표본 유효 분해능: ~1 nm (시료 준비 문제, contrast 한계 등)

수차 (Aberration)
렌즈를 통과한 빛이 한 점에 모이지 못해 색이나 상이 왜곡되는 현상
- 색수차(Chromatic Aberration): 전자빔의 에너지 불균일성(파장 차이) 때문 발생
- 구면수차(Spherical Aberration): 렌즈 중심과 주변부를 통과하는 전자의 초점 차이 → 주변부 전자의 굴절 과다
전자 산란과 대비 형성
생체 구성 원소(C, H, O, N)는 원자번호 낮음 → 전자 산란 적음 → 대비 낮음(밝게 나타남) → 중금속 염색 필요: 핵전하 ↑ → 전자 산란 ↑ → 이미지 대비 ↑
BF (Bright Field, 명시야): 직접 통과 전자 이용 → 중금속 부위가 어둡게 보임 (생체 시료에서 가장 흔히 사용) DF (Dark Field, 암시야): 산란된 전자만 이용 → 중금속 부위가 밝게 보임 (BF와 대비 반대)
생물학적 시료 준비
→ Cryo EM_Biological Specimen Preparation for Electron Microscopy
전자빔은 매우 높은 에너지 + 진공 상태 → 살아있는 세포 관찰 불가
Conventional TEM 준비
고정(Fixation) → 탈수(Dehydration) → 포매(Resin Embedding) → 초박절편(Ultramicrotomy) → 염색
- 고정: Glutaraldehyde (단백질 고정) + Osmium tetroxide (OsO₄) (지질·세포막 보존, 전자밀도 증가)
- 탈수: 물을 유기용매로 대체 (물은 진공에서 증발, 레진과 섞이지 않음)
- 포매: 레진 침투 → 블록화
- 초박절편: ultramicrotome으로 수십 nm 두께로 절단 → metal grid 위에 올려 관찰
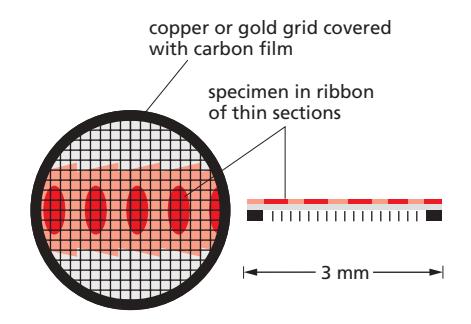
- 대비 염색: uranyl acetate / phosphotungstic acid / ammonium molybdate (원자번호 높은 물질)
Negative Staining (음성 염색)
→ positively stained vs negatively stained

- 염색이 내부로 들어가지 않고 외부에만 존재
- 외부만 전자 산란이 많아져 어둡게 보임 → 시료 윤곽이 밝게 보임
- 주로 분리된 단백질, 바이러스 등 macromolecule 관찰에 사용
Cryo EM
- Vitrification (급속 동결): 물을 얼음 결정이 아닌 비결정질(유리질) 형태로 유지 → 구조 왜곡 최소화
- 이로써 전자빔에 의해 얼음이 녹지 않음
- Cryo-sectioning: 두꺼운 세포를 **집속 이온 빔(FIB)**으로 깎아내려 판 모양(lamella)으로 만든 후 관찰 (염색 없이)
3D 재구성
Serial sectioning → 3D 재구성: 다른 깊이의 절편 이미지들 → 컴퓨터로 쌓아 3D 구조 재구성
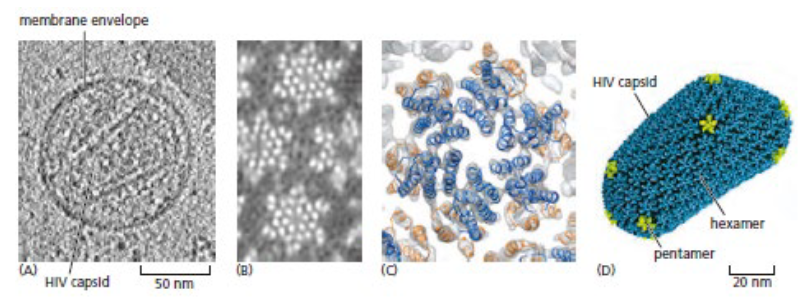
Single Particle Reconstruction (단일 입자 재구성):
→ Single Particle Reconstruction
- 전자빔이 생체분자를 손상시켜 한 분자에서 얻는 전자 신호가 제한적
- 따라서 단일 이미지로는 높은 해상도의 구조를 얻기 어려움
- 단일 입자 재구성: 동일한 분자를 낮은 전자 밀도로 매우 많이 촬영 → 정렬(alignment) + 평균화(averaging) → 평균 이미지 생성 → SNR 향상
- 예: HIV 캡시드 내부 단백질 구조 → **216개의 6량체(hexamer, 파란색)**와 12개의 5량체(pentamer, 노란색)
SEM (Scanning Electron Microscope)
→ SEM(Scanning Electron Microscope)
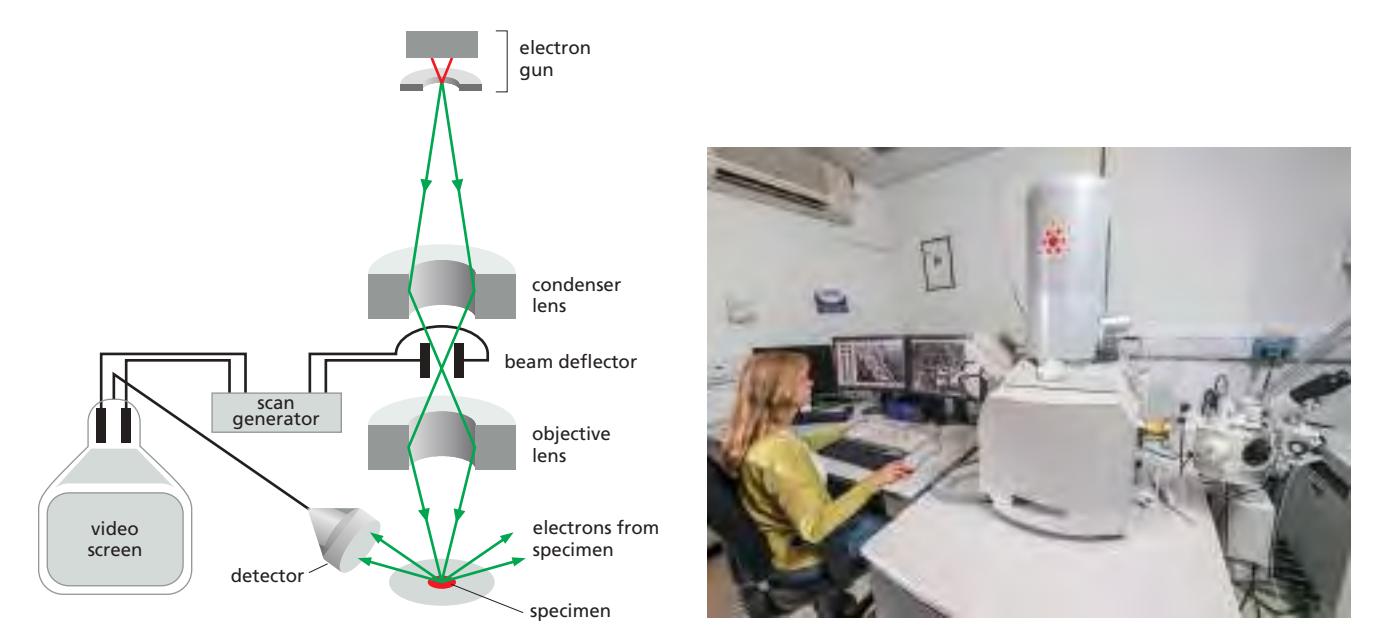
시료와 전자빔 사이에서 일어나는 ‘상호작용’을 데이터화하여 시각화
- 후방 산란전자(Backscattered electron, BSE): 1차 전자가 핵에 의해 산란되어 나온 전자
- 이차전자(Secondary electron, SE): 시료 표면에서 방출 → 표면 확인
- 내부 구조 관찰 불가 → 주로 표면 3D 이미지 생성
- 해상도: ~10 nm (standard SEM)
현미경 종합 비교표
| 현미경 | 분해능 | 살아있는 세포 | 주요 용도 |
|---|---|---|---|
| 명시야(Bright-field) | ~200 nm | 고정/생 모두 | 염색된 조직 |
| 암시야(Dark-field) | ~200 nm | 가능 | 산란광 관찰 |
| 위상차(Phase-contrast) | ~200 nm | 가능 (No staining) | Live cell imaging |
| 형광(Fluorescence) | ~200 nm | 가능 | 특정 분자 표지 |
| Confocal | ~200 nm | 가능 | 3D 광학 절편 (150 μm) |
| Multiphoton | ~200 nm | 가능 | 깊은 조직 (500 μm) |
| TIRF | ~200 nm | 가능 | 세포막 근처 (200 nm) |
| SIM | ~100 nm | 어려움 | 초고해상도 |
| STED / SMLM | ~20 nm | 어려움 | 초고해상도 |
| TEM | ~0.05 nm== | 불가 | 내부 초미세 구조 |
| SEM | ~10 nm | 불가 | 표면 3D 이미지 |
| Cryo EM | ~nm | 불가 | 단백질 3D 구조 |
| AFM | 0.1 nm (수직) | 가능 | 표면·물성 측정 |
관련 노트 모음
광학현미경
- optical microscope — 기본 원리
- limit of resolution(분해능) — 분해능
- diffraction limit — 회절한계
- contrast(대비) — 대비 형성 3가지 방식
- phase contrast microscopy(위상차 현미경) — 위상차 현미경
- 4 types of microscopy(시험예시 이미지) — 시험 예시 이미지
- PSF(point spread function) — PSF
- deconvolution — 이미지 복원
형광현미경
- 000_Fluorescence Microscopy(형광현미경) — 형광현미경 전반
- Immunofluorescence Staining — 항체 기반 형광 염색
- GFP(Fluorescent Protein Tagging) — GFP 및 융합 단백질
- Confocal Microscope — 공초점 현미경
- Multiphoton Microscopy — 다광자 현미경
- TIRF(Total Internal Reflection Fluorescence Microscopy) — TIRF
- Fluorescent Biosensors for Cell Signaling — 형광 바이오센서
Protein Dynamics
- 000_Protein Dynamics in vivo MOC — 개요
- FRET — 형광 공명 에너지 전달
- Photoactivation — 광활성화
- FRAP — 광표백 후 회복
- Aequorin=Light Emitting Indicator — Ca²⁺ 농도 측정
초고해상도
- SIM(Structured Illumination Microscopy) — SIM
- STED(Stimulated Emission Depletion) — STED
- SMLM (Single-Molecule Localization Microscopy) — SMLM/PALM
전자현미경
- TEM(Transmission Electron Microscope) — TEM
- SEM(Scanning Electron Microscope) — SEM
- Cryo EM_Biological Specimen Preparation for Electron Microscopy — Cryo EM 및 시료 준비
- positively stained vs negatively stained — 양성/음성 염색
- Single Particle Reconstruction — 단일 입자 재구성
기타
- Sectioning — 조직 고정 및 절편
- AFM(Atomic Force Microscopy) — 원자력 현미경
- 현미경 별 관찰 규모 — 스케일 개요